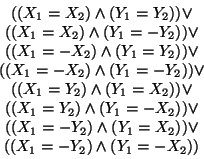Simple Lattice Model of Protein Folding and the Inverse Folding Problem
The purpose of this work is to provide the description and the results of a
simple lattice model for protein folding. We also describe a recent
polynomial approach to the inverse protein folding problem as proposed by
J. Kleinberg.
The lattice model is a simplified two-dimensional version of the 3-D model
suggested by researchers at the NEC Center in Princeton. An interesting
application described herein is to apply Kleinberg's inverse protein
folding approach to the main configurations of proteins in the 2-D lattice
model.
This web page contains:
Given a
3x3x3 cube with 27 internal mini-cubes, consider a Self-Avoiding Path
(SAP) made-up of 27 edges of the mini-cubes(see Figure 1). A node in the SAP is one of the
vertices of the mini-cubes and an edge corresponds to an edge of a
mini-cube. A SAP does not have
intersecting edges (i.e., edges that are shared or touch another edge in the
path). In other words, no two nodes in a SAP have the same coordinates (i , j,
k). It is assumed that an SAP starts at the point (0, 0, 0).
It
can be shown that there are about 50,000 such different SAP's that are not obtained
from another SAP by symmetry considerations. Let us define as Untouching
Neighboring Nodes (UNN) any two nodes in an SAP that are not in the same
edge of the path but are neighboring in the sense that they belong to an edge
in a mini-cube.
Fig. 1 The Lattice Model
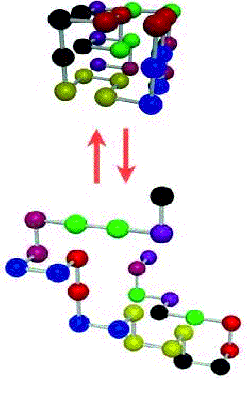
The figure shows a 3x3x3 lattice and a Self-Avoiding Path (SAP).
Now consider all binary strings of length 27. There are
227 such strings. Notice that this number of strings is much larger than the
number of SAPs in the original cube.
Each of the ones and zeros in a binary string S will
correspond to an aminoacid . Aminoacids are roughly classified into two types:
Hydrophopic (H) or Polar (P). Hydrophobicity is a property of an aminoacid that
specifies a preference to be placed
in the "inside" of the protein. Alternatively, a polar aminoacid
preferentially occupies a position in the surface of the protein.
Intuitively,
the approximate protein folding problem consists of determining, for each
binary string S (also called an HP string), the "optimal" SAP, that is, the one
that places "most" of its H elements in the interior part of the cube, and
"most" of its P elements in the surface of the original cube. The words
"optimal" and "most" are quantitatively defined in the sequel.
A
pair of UNN nodes in the cube can be of three types: HH, PP, and HP, since HP
and PH represent the same pair. We
now define an energy function E(P,S) where P is a SAP and S is a 27
digit binary string representing a protein. Given a string S and a SAP path P, E is computed as
follows:
Let
i and j be the indices of elements in a string S such that they correspond to a
pair of UNN nodes in the cube. Then:
where P(i,j) = 3 if i and j are both hydrophobic; P(i,j) = 1 if
i and j are one hydrophobic and one polar; P(i,j) = 0 for all other pairs.
In
the lattice model one considers a given binary string S and exhaustively
determines, from all SAPs, the one that evaluates to a minimal energy E. A
question arises: What is the distribution of the S's within all SAP's? The
researchers of NEC at Princeton [1] considered that problem and solved it
by exhaustive search for each of the 27 digit binary string.
An
important remark is in order: if a given string optimally matches two more SAPs
then it is reasonable to disregard it, since, in nature, a sequence of aminoacids folds predominantely to a unique configuration.
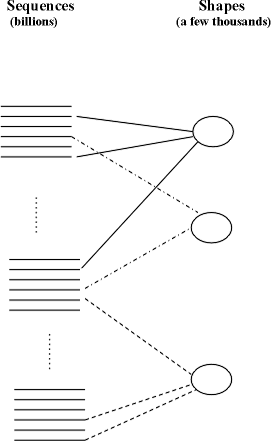
There
are a large number of strings S that match a given optimal SAP. This means that
many sequences of aminoacids acquire the same shape when folded. This fact is
validated in laboratory experiments. In
humans, there may be 100,000 valid sequences of aminoacids corresponding to
genes, and it is estimated that there may be only a few thousand shapes that
those proteins fold into (see Figure 2). Each of those 100,000 sequences contains hundreds of
aminoacids. In nature, each of the 20 existing aminoacids has a different
degree of polarity and hydrophobicity specified by a real number between 0 and 1.
Fig. 2
HP Sequence Mapping to Shapes
Instead of a 3x3x3 lattice, we used a 4x4 2-D lattice model.
Figure 3
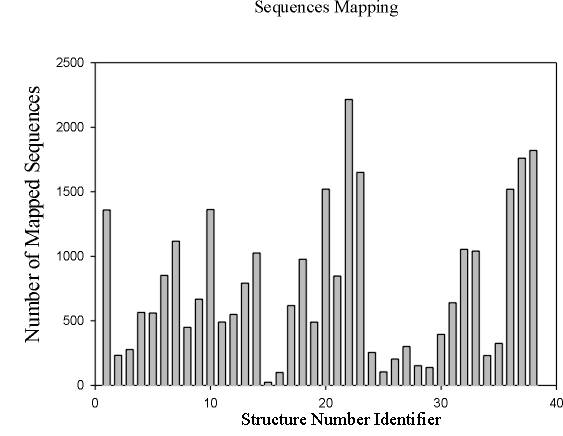
shows the SAPs for such a model after symmetry elimination. There are totally 65,536 HP sequences that can be mapped onto the 4x4 lattice. We mapped those sequences to the 38 SAPs using a NEC-like approach.
Figure 4 shows the distribution of the mapping. Note that the SAP numbered
22 is the one that corresponds to the most
frequent optimal shape of all the 65,536 HP strings considered in the 2-D model.
Fig. 3
The 38 SAPs After the Symmetry Elimination
structure 1:
*--* *--*
| | | |
* * * *
| | | |
* * * *
| | | |
* *--* *
-----------------
structure 2:
*--* *--*
| | | |
* * * *
| | |
* * *--*
| | |
* *--*--*
-----------------
structure 3:
*--* *--*
| | |
* * *--*
| | |
* * *--*
| | |
* *--*--*
-----------------
structure 4:
*--* *--*
| | | |
* * * *
| | | |
* * * *
| | |
* *--*--*
-----------------
structure 5:
*--* *--*
| | | |
* * * *
| | | |
* *--* *
| |
* *--*--*
-----------------
structure 6:
*--* *--*
| | |
* *--* *
| | |
* *--* *
| | |
* *--*--*
-----------------
structure 7:
*--* *--*
| | | |
* *--* *
| |
* *--*--*
| |
* *--*--*
-----------------
structure 8:
*--* *--*
| | | |
* *--* *
| |
* *--* *
| | |
* *--*--*
-----------------
structure 9:
*--* *--*
| | | |
* *--* *
| |
* *--* *
| | | |
* * *--*
-----------------
structure 10:
*--*--*--*
| |
* *--*--*
| |
* * *--*
| | | |
* *--* *
-----------------
structure 11:
*--*--*--*
| |
* *--*--*
| |
* * *--*
| | |
* *--*--*
-----------------
structure 12:
*--*--*--*
| |
* *--*--*
| |
* *--*--*
| |
* *--*--*
-----------------
structure 13:
*--*--*--*
| |
* * *--*
| | |
* * *--*
| | |
* *--*--*
-----------------
structure 14:
*--*--*--*
| |
* *--* *
| | | |
* * *--*
| |
* *--*--*
-----------------
structure 15:
*--*--*--*
| |
* *--* *
| | | |
* * * *
| | |
* *--*--*
-----------------
structure 16:
*--*--*--*
| |
* *--* *
| | |
* *--* *
| | |
* *--*--*
-----------------
structure 17:
*--*--*--*
| |
* *--* *
| | | |
* * * *
| | | |
* * *--*
-----------------
structure 18:
*--*--*--*
| |
*--*--* *
|
*--* *--*
| | |
* *--*--*
-----------------
structure 19:
*--*--*--*
| |
*--* *--*
|
*--* *--*
| | |
* *--*--*
-----------------
structure 20:
* *--*--*
| | |
*--* *--*
|
*--* *--*
| | |
* *--*--*
-----------------
structure 21:
*--*--*--*
| |
*--*--* *
| |
*--* * *
| | |
* *--*--*
-----------------
structure 22:
*--*--*--*
| |
*--* *--*
| |
*--* *--*
| |
* *--*--*
-----------------
structure 23:
*--*--*--*
| |
*--*--* *
| |
*--*--* *
| |
* *--*--*
-----------------
structure 24:
*--*--*--*
| |
*--* *--*
| |
* * * *
| | | |
*--* *--*
-----------------
structure 25:
*--*--*--*
| |
*--* *--*
| |
* * *--*
| | |
*--* *--*
-----------------
structure 26:
*--*--*--*
| |
*--* * *
| | |
* * * *
| | | |
*--* *--*
-----------------
structure 27:
*--*--*--*
| |
* *--* *
| | |
* * * *
| | | |
*--* *--*
-----------------
structure 28:
*--*--*--*
| |
*--*--* *
|
* *--* *
| | | |
*--* *--*
-----------------
structure 29:
*--* *--*
| | | |
* *--* *
|
* *--* *
| | | |
*--* *--*
-----------------
structure 30:
*--*--*--*
| |
*--*--* *
| |
* *--* *
| | |
*--* *--*
-----------------
structure 31:
*--*--*--*
| |
*--* *--*
|
* *--*--*
| |
*--*--*--*
-----------------
structure 32:
*--*--*--*
| |
* *--*--*
|
* *--*--*
| |
*--*--*--*
-----------------
structure 33:
*--*--*--*
| |
*--* *--*
| |
* * *--*
| |
*--*--*--*
-----------------
structure 34:
*--*--*--*
| |
*--* * *
| | |
* *--* *
| |
*--*--*--*
-----------------
structure 35:
*--*--*--*
| |
*--*--* *
| |
* *--* *
| |
*--*--*--*
-----------------
structure 36:
*--*--*--*
| |
*--* *--*
|
*--* *--*
| |
*--*--*--*
-----------------
structure 37:
*--*--*--*
| |
*--*--* *
| |
*--* * *
| |
*--*--*--*
-----------------
structure 38:
*--* *--*
| | | |
* * * *
| | | |
* * * *
| |
*--*--*--*
-----------------
Fig. 4 Number of HP Strings Mapped to SAPs
We now define the inverse protein folding problem. Given a SAP or
a structure 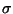 in space, determine all the
strings S that have as the structure minimizing the energy E.
in space, determine all the
strings S that have as the structure minimizing the energy E.
Kleinberg
in [2] proposes a polynomial approximate algorithm:
given
a structure specified by a sequence of straight line segments in space ,
determine the set of HP strings that
are potential candidates for having as the folded structure. Therefore, he provides an approximate
solution to the inverse protein folding problem. Kleinberg uses a Fitness
Function 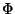 instead of the previously defined Energy function.
instead of the previously defined Energy function.
is defined as:
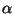 and
and 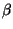 are constants; the sum of d(i,j) correspond
to the distances between space coordinates of the nodes, and si corresponds
to the nodes that are in the surface of the given . SH denotes the set of numbers i such that the ith position of the sequence S is H (Hydrophobic).
are constants; the sum of d(i,j) correspond
to the distances between space coordinates of the nodes, and si corresponds
to the nodes that are in the surface of the given . SH denotes the set of numbers i such that the ith position of the sequence S is H (Hydrophobic).
In what follows we summarize how one would make practical usage of the inverse
protein folding approach. Assume that given a shape one could
determine all the HP strings corresponding to that
.
Kleinberg shows how one
can determine one such HP string and he then discusses how one could
approach the problem of finding the set of strings that would also fold into
.
He considers the set of HP strings obtainable from the optimal string by
the change of one amino acid type, i.e., an H being changed into a P, or a P
being changed into an H.
Kleinberg showed that such an ensemble of strings may not have a continuous
space representation. This means that if a string Si is transformed -- by a
change of one aminoacid type into a string Sj -- and afterwards to a string
Sk also by a single change, then it may occur that Si and Sk are optimally
foldable into
whereas Sj is not. In other words the space of solutions
of strings matching is not convex or continuous. Kleinberg
calls this set of all the Si having as optimal shape, the landscape
L of
It turns out that determining L is itself an exponential problem since it may
contain a very large number of strings. It would be therefore desirable to construct
a recognizer R (say, an automaton), capable of determining if a given
string S is recognized by R. This is an open problem that could be studied by
machine learning approaches (or grammar inference techniques) using the model
described in this work.
Given the pairs
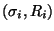 for a large number of different shapes
for a large number of different shapes
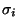 ,
known to exist in nature, then the search for a shape
,
known to exist in nature, then the search for a shape 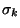 corresponding to
a given string S would proceed as follows. Attempt to recognize S by each of
the recognizers Ri. Assume it is accepted by the
k-th recognizer Rk. If that is the case,
is the most likely structure
for S.
corresponding to
a given string S would proceed as follows. Attempt to recognize S by each of
the recognizers Ri. Assume it is accepted by the
k-th recognizer Rk. If that is the case,
is the most likely structure
for S.
Let us now return to the problem of finding one HP string that optimizes a given structure
.
Kleinberg shows that the problem
can be reduced to maximal flow-minimum cut of a graph directly obtainable from
. In Figure 5, we show the graph obtained from the structure numbered 22. The vertex set V of the graph G consists of s, t, a vertex vi for each of the residue position i = 1,2,...,16 in the target shape, and a vertex uij for each pair of residue positions i,j for each i<j-2 and g(dij)>0. The edge set E of G consists of an edge (s,uij) for each vertex uij, and edge (vi,t) for each vertex vi which has a non-zero solvent-exposed contact surface area si and edges (uij,vi) and (uij,vj) for each vertex uij. We assign a capacity of
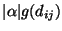 to the edge (s,uij), a capacity of
to the edge (s,uij), a capacity of
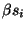 to the edge (vi,t), and a capacity of
B+1, where B is
to the edge (vi,t), and a capacity of
B+1, where B is
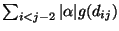 , to all edges of the form (uij, vi) and (uij,vj).
, to all edges of the form (uij, vi) and (uij,vj).
Figure 5 The Graph for Kleinberg's Algorithm for the Structure Numbered 22
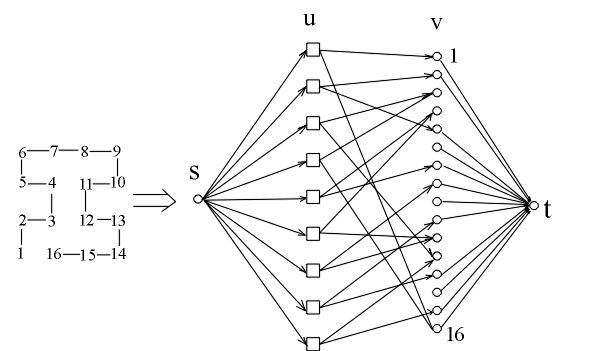
Notice that there are 9 pairs of nodes(UNNs)
1 1- 16
2 2- 5
3 3- 12
4 3- 16
5 4- 7
6 4- 11
7 8- 11
8 10-13
9 12-15
The nodes 3,4,11 and 12 are internal nodes and therefore there are no edges linking them to the sink node t.
The Kleinberg method was applied to the 38 SAPs of the 2-D lattice model. That
method determines one HP sequence for each SAP that minimizes the fitness function value defined above.
Note that for some of the SAPs, the sequences found by Kleinberg's method are also mapped by
the NEC standard while others are not. (See Table 1) The values for the constants
and
where taken to be -2 and 0.1 respectively. (Note in the program, the values are set to be -20 and 1. Thus the program computes 10 x . However this won't change the sequence yielding the minimal fitness value for the given structure.) It may be worthwhile to determine the values to maximize the number of 'Yes' entries in Table 1.
Table 1. Result of Kleinberg Method on 2-D Model
|
Structure
|
Number of Mapped Sequences by NEC Method
|
Sequence Found by Kleinberg Method |
NEC and Kleinberg Method Map the Same Sequence
|
|
1
|
1358
|
HHHPHHHHHHHHPHHH
|
No
|
|
2
|
232
|
HPHHHHPHHHHHPHHH
|
No
|
|
3
|
276
|
HPHHHHPHHHHHPHHH
|
No
|
|
4
|
564
|
HHHPHHPHHHHHPHHH
|
No
|
|
5
|
560
|
HHPHHPHHHHHHPHHH
|
Yes
|
|
6
|
851
|
HPHHPHHHHHHHPHHH
|
Yes
|
|
7
|
1116
|
HHHHHHHPHHHHPHHH
|
No
|
|
8
|
449
|
HHHHPHHPHHHHPHHH
|
No
|
|
9
|
667
|
HHHHPHHPHHHHPHHH
|
No
|
|
10
|
1361
|
HHPHHHHHHPHHPHHH
|
No
|
|
11
|
490
|
HHPHHHHHHPHHPHHH
|
No
|
|
12
|
549
|
HHPHHHHHHPHHPHHH
|
No
|
|
13
|
791
|
HHHHPHHHHPHHPHHH
|
Yes
|
|
14
|
1025
|
HHHHHHHHHPHHPHHH
|
No
|
|
15
|
23
|
HHHHHHPHHPHHPHHH
|
No
|
|
16
|
98
|
HHHHHHPHHPHHPHHH
|
No
|
|
17
|
617
|
HHHHHHPHHPHHPHHH
|
No
|
|
18
|
976
|
HPHHPHHHHHPHHPHH
|
No
|
|
19
|
489
|
HHPHHPHHHHPHHPHH
|
No
|
|
20
|
1520
|
HHPHHPHHHHPHHPHH
|
No
|
|
21
|
846
|
HHHHPHHPHHPHHPHH
|
No
|
|
22
|
2214
|
HHPHHHHPHHPHHHHH
|
Yes
|
|
23
|
1650
|
HHPHHPHHPHHHHHHH
|
Yes
|
|
24
|
253
|
HPHHHHPHHPHHHHPH
|
No
|
|
25
|
103
|
HPHHHHPHHPHHHHPH
|
No
|
|
26
|
203
|
HHHPHHPHHPHHHHPH
|
No
|
|
27
|
300
|
HPHHPHHPHHHHHHPH
|
No
|
|
28
|
151
|
HHHPHHPHHPHHHHPH
|
No
|
|
29
|
137
|
HPHHHHPHHPHHHHPH
|
No
|
|
30
|
394
|
HPHHPHHPHHHHHHPH
|
No
|
|
31
|
639
|
HHPHHPHHHHHPHHPH
|
Yes
|
|
32
|
1053
|
HPHHPHHHHHHPHHPH
|
Yes
|
|
33
|
1040
|
HHHPHHPHHHHPHHPH
|
Yes
|
|
34
|
230
|
HHHHHPHHPHHPHHPH
|
No
|
|
35
|
325
|
HHHHHPHHPHHPHHPH
|
No
|
|
36
|
1519
|
HHPHHPHHHHPHHPHH
|
Yes
|
|
37
|
1760
|
HHHHPHHPHHPHHPHH
|
Yes
|
|
38
|
1819
|
HHHPHHPHHPHHPHHH
|
Yes
|
- Attempt to find the recognizers for
the landscapes of each of the 38 SAPs of the model
(The strings matching each of them can be obtained by the enclosed program.).
This attempt should be considered successful if the intersection of any two
recognizers is empty, that is, no string is accepted by any two of the
recognizers. A recognizer could be a FSG grammar inferred from the known
strings matching a given SAP.
- Attempt to find alignment weights for each of the SAPs of the model. In other
words the recognizer for testing if a string corresponds to one of the SAPs is
determined by a non-gapped alignment determining a score for comparing a
candidate HP string S with the optimal HP string determined by Kleinberg's
method for each of the SAPs in the model.
- Check if an existing short protein has the shape SAP suggested by the model.
- Apply Kleinberg
landscape results to the families of strings corresponding to each SAP.
- Is it valid to subdivide a bigger lattice (say 8 x 8) as made up
of 4 sets of 4 x 4 lattices?
- Check if the following is true: 2 widely different strings match to the same
SAP? (by widely different one means having a totally different ratios of Hs and
Ps) Alternatively determine if two similar strings (say differing by one amino acid type H or P) belonging to a given SAP are in the
same family?
- Kleinberg states that one can generalize his model to consider aminoacids
having given degrees of polarity and hydrophobicity. Is that applicable to the
simple model?
- 1
- Hao Li, Robert Helling, Chao Tang, and Ned Wingreen. (1996) ``Emergence of
Preferred Structures in a Simple Model of Protein Folding'' Science,1996(273):666-669
- 2
- Jon M. Kleinberg. (1999) ``Efficient Algorithms for Protein Sequence Design
and the Analysis of Certain Evolutionary Fitness'' Proc. 3rd ACM RECOMB Intl. Conference on Computational Molecular Biology, 1999.
- 3
- G. Mauri, A. Piccolboni, G. Pavesi. (1999) ``Approximation Algorithms for Protein Folding Prediction.'' Poster Session, ACM Recomb 99 Conference, Lyons, France 1999.
- 4
- H.S. Chan, K.A. Dill. (1993) ``The Protein Folding Problem'' Physics Today 1993:24-32
- 5
- W.H. Hart, S.C. Istrail. (1996) ``Protein Folding in the Hydrophobic-Hidrophilic Model within 3/8 of Optimal.'' Journal of Computaional Biology 1996
Protein Folding in the Generalized Hydrophobic-Polar Model on the Triangular Lattice
Approximate Protein Folding in the HP Side Chain Model on Extended Cubic Lattices
Local Rules for Protein Folding on a Triangular Lattice and Generalized Hydrophobicity in the HP Model
Exploring the Fitness Landscapes of Lattice Proteins
Germ�n Terrazas's Protein Data
LEDA and LEDA Library
Appendix
1. Computation of Self-Avoiding Paths (SAP)
We determined the SAPs(Self-Avoiding Path) of a 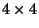 2-D lattice using a
Prolog program. ``sap(X,Y)'' determines the SAP and the pairs of contacting
residues in the SAP. ``sappath(X)'' is the clause that finds the SAP. It
first makes a list of residues representing the path. Then it sets the constraints
for the path. There are two constraints for a self-avoiding path. One is set
by ``rightsucc(X)'' which ensures that residue i and residue i+1 are neighbors.
This is satisfied by applying the constraint that only one of the
2-D lattice using a
Prolog program. ``sap(X,Y)'' determines the SAP and the pairs of contacting
residues in the SAP. ``sappath(X)'' is the clause that finds the SAP. It
first makes a list of residues representing the path. Then it sets the constraints
for the path. There are two constraints for a self-avoiding path. One is set
by ``rightsucc(X)'' which ensures that residue i and residue i+1 are neighbors.
This is satisfied by applying the constraint that only one of the
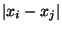 or
or
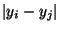 is equal to 1. The second constraint for
the SAP is self-avoidance, which means there is no crossing of a path with itself. This
is set by ``selfavoid(X)'', which ensures that no two residues can occupy
the same location. The ``allcontact(X,Y)'' actually does a linear search
for each residue to find its neighbors and place the result in ``Y''.
is equal to 1. The second constraint for
the SAP is self-avoidance, which means there is no crossing of a path with itself. This
is set by ``selfavoid(X)'', which ensures that no two residues can occupy
the same location. The ``allcontact(X,Y)'' actually does a linear search
for each residue to find its neighbors and place the result in ``Y''.
2. Symmetry Detection
Among all the SAPs, some are symmetrical to the others. For example, the points
P(X,Y) and P(X,-Y) are symmetric. There are 8 types of symmetries.
If
P1(X1,Y1),
P2(X2,Y2) are two positions in
the two symmetrical structures, each of the following clauses describes a condition
for one type of symmetry.
In the program, the function ``IsSymmetry'' tests whether two structures
are symmetric. It does so by testing if the two structures satisfy one
of above conditions.
3. Mapping Sequences to Structures Using NEC Energy Formula
Given a sequence, the energy is computed for each shape. The energy is defined
as follows: For each pair of contacted residues, if the two residues
are both hydrophobic they contribute -3 to the energy. If one of the residue
is hydrophobic, they contribute -1 to the energy. If none of the residues is
hydrophobic, they contribute 0 to the energy.
For each sequence, the energy is computed over all the structures. If there
is only one structure that gives minimal NEC energy, this sequence is mapped
to the structure. After doing this mapping for all sequences, for each structure,
we have a set of sequences that map to the structure. The number of mapped sequences
over structure is plotted in Figure 4.
In the program, the function ``ComputeNECEnergy'' computes the energy
as described above. The function ``FindMinValue'' function finds the minimal
energy value over all the shapes to a particular sequence. The function ``FindShape''
finds the only one structure that yields this minimal value and map the sequence
to the structure by adding the sequence to the structure's sequence list using the
function ``AddRecord''. The number of mapped sequences to each structure
is counted and recorded.
4. Find Sequence for Structures with Kleinberg Fitness Value
Kleinberg fitness value is defined by
g(dij) = 1 if both residue i and j are hydrophobic and are
neighbors; 0 otherwise.
Si = 0 if the residue is not on the surface;1 if the residue
is on the surface but not at the corner; 2 if the residue is at the corner.
We use both a brute force and a network mini_cut algorithm to determine the sequences
that give the minimal Kleinberg fitness value to a given structure. The brute
force approach simply considers all the sequences and computes the Kleinberg
fitness value for each structure. Then it finds the sequence that gives the
minimal fitness value to each structure. Kleinberg used a polynomial algorithm to
find the sequence yielding the minimal fitness value. Basically, a weighted directed graph
is built according to the given structure. Then the mini_cut of the graph partitions
the graph into two parts. The nodes in one part represent hydrophobic residues
while the nodes in the other represent polar residues. The graph network mini_cut algorithm is described in Kleinberg's paper.
In the program, we first build the graph according to Kleinberg's method in function``MakeGraph''.
Then we determine the max_flow of the graph using the algorithm library LEDA. This
also give us the flow for each edge. In function ``MarkGraph'' we find
the mini_cut of the graph by a marking method. The method can be described as follows:
Mark the source. Examine all the out-going edges from the
source. If the flow through the edge is larger than zero and less than the capacity
of the edge, mark the node reachable from that edge. The source is now processed.
Then select an unprocessed node from the set of all marked nodes and do the same operation
until the set of marked nodes remains unchanged. The graph is partitioned into marked
and unmarked two parts after the procedure.
The residues whose representing nodes are in the marked set are hydrophobic
and the others are polar.
SAP Computation in Prolog:
/*
* This program determines the self avoid path of a 4x4 2-D lattice.
* The query sap(Path, Contacts) to get the results. The 'Path'
* will be a list of the residues formated as
* [residue(1,X1,Y1), residue(2,X2,Y2), ...residue(16, X16,Y16)]
* Xi and Yi are x,y coordinations of the amino acid.
* The 'Contacts' will be a list of all the contacts in that
* configuration in the format of:
* [c(1,4), c(2,3), ...c(16,7)]
*/
sap(Path, Contacts):-
sappath(Path),
allcontact(Path, Contacts).
sappath(Path):-
length(Path),
rightsucc(Path),
selfavoid(Path),
labeling(Path).
length([residue(1,X1,Y1),residue(2,X2,Y2),residue(3,X3,Y3),residue(4,X4,Y4),
residue(5,X5,Y5),residue(6,X6,Y6),residue(7,X7,Y7),residue(8,X8,Y8),
residue(9,X9,Y9),residue(10,X10,Y10),residue(11,X11,Y11),residue(12,X12,Y12),
residue(13,X13,Y13),residue(14,X14,Y14),residue(15,X15,Y15),residue(16,X16,Y16)]).
rightsucc([]).
rightsucc([residue(I,X,Y)]).
rightsucc([residue(I1,X1,Y1), residue(I2,X2,Y2)|Rp]):-
{abs(X1-X2)+abs(Y1-Y2)=1},
rightsucc([residue(I2,X2,Y2)|Rp]).
selfavoid([]).
selfavoid([residue(I,X,Y)|Rp]):-
avoid(residue(I,X,Y), Rp), selfavoid(Rp).
avoid(residue(I,X,Y), []).
avoid(residue(I,X,Y), [residue(Is,Xs,Ys)|Rp]):-
{abs(X-Xs)+abs(Y-Ys) =\= 0},
avoid(residue(I,X,Y), Rp).
labeling([]).
labeling([residue(I,X,Y)|T]):-
member(X,[1,2,3,4]),
member(Y,[1,2,3,4]),
labeling(T).
member(X,[X|_]).
member(X,[_|T]):-
member(X,T).
allcontact([],[]).
allcontact([residue(I,X,Y)],[]).
allcontact([residue(I,X,Y), residue(It,Xt,Yt)|Rp], Res) :-
contact(residue(I,X,Y), Rp, Cont),
allcontact([residue(It,Xt,Yt)|Rp], AllCont),
append(Cont, AllCont, Res).
contact(residue(I,X,Y), [], []).
contact(residue(I,X,Y), [residue(Is,Xs,Ys)|Rp], Res):-
{abs(X-Xs)+abs(Y-Ys) = 1},
contact(residue(I,X,Y), Rp, Conts),
append([c(I,Is)], Conts, Res).
contact(residue(I,X,Y), [residue(Is,Xs,Ys)|Rp], Res):-
{abs(X-Xs)+abs(Y-Ys) =\= 1},
contact(residue(I,X,Y), Rp, Res).
append([], X, X).
append([H|T], X, [H|S]):-
append(T,X,S).
Sequence Mapping and Kleinberg Method in C:
#include <stdio.h>
#include <stdlib.h>
#include <string.h>
#include <math.h>
#include <LEDA/graph.h> //The LEDA library
#include <LEDA/graph_alg.h>
#define HHENERGY 3
#define HPENERGY 1
#define SEQLENGTH 16 //This is a 16-node lattice
#define SEQMAX 65535
#define Alpha -20 //The alpha parameter in Kleinberg function
#define Belta 1 //The beta parameter in Kleinberg function
////////////////////////////////////////////////////////////////////
// The data structures:
////////////////////////////////////////////////////////////////////
/*****************************************************
* This is the sequence
*****************************************************/
char Seq[16] = {0,0,0,0,
0,0,0,0,
0,0,0,0,
0,0,0,0};
/******************************************************
* This represents a list of sequence which maps to
* a particular structure
******************************************************/
struct seqlist;
struct seqlist {
struct seqlist *next;
unsigned int sequence;
unsigned int energy;
};
/*********************************************************
* This represents a list of all the neighboring residues
* in a particular structure
*********************************************************/
struct contact;
struct contact {
struct contact *next;
int i;
int j;
};
/*****************************************************************
* This represents a list of the structures. Each
* one contains the following information:
* 1. The index of the structure
* 2. An array of residues in the structure and their coordination
* 3. A list of neighboring residues
* 4. A list of sequences
* 5. the number sequences mapped to the structure
* 6. the minimal energy
*****************************************************************/
struct shapeset;
struct shapeset {
int index;
int seqcount;
int kmapseq;
int Xs[SEQLENGTH]; //An array of residues X coordinates
int Ys[SEQLENGTH]; //An array of residues Y coordinates
double energy;
struct shapeset * next;
struct contact * clist; //A list of neighboring residues
struct seqlist * slist; //A list of sequences
};
////////////////////////////////////////////////////////////////////
// The util functions for read in structures and printing out result
// and some manipulations functions for the above data structure
////////////////////////////////////////////////////////////////////
unsigned int ReadLine(FILE* f, char b[], size_t size)
{
unsigned int i = 0;
char c;
c=fgetc(f);
while(c!='\n' && c != EOF && i<size-1) {
b[i++] = c;
c=fgetc(f);
}
b[i] = '\0';
return i;
}
int GetResidues(char rds[], struct shapeset * element)
{
const char delimiters[] = "[(),";
char* token;
int pos = 0;
int index;
token = strtok (rds, delimiters);
index = atoi(token);
token = strtok (NULL, delimiters);
while (token) {
if (token[0] != ' ' && token[0] != ']') {
element->Xs[pos] = atoi(token);
token = strtok (NULL, delimiters);
element->Ys[pos] = atoi(token);
pos++;
}
token = strtok (NULL, delimiters);
}
return index;
}
void GetContact(char cts[], struct shapeset * element)
{
const char delimiters[] = "(),";
char* token;
int pos = 0;
struct contact * nct;
token = strtok (cts, delimiters);
token = strtok (NULL, delimiters);
while (token) {
if (token[0] != ' ' && token[0] != ']') {
nct = (struct contact *)
calloc(1, sizeof(struct contact));
nct->i = atoi(token);
token = strtok (NULL, delimiters);
nct->j = atoi(token);
pos++;
if ( !element->clist ) element->clist = nct;
else {
struct contact * tail = element->clist;
while (tail->next) tail = tail->next;
tail->next = nct;
}
}
token = strtok (NULL, delimiters);
}
}
void ReadConformation( char * filename, struct shapeset **set )
{
FILE * f = fopen(filename, "r");
char line[256];
int i;
struct shapeset * nset;
i = ReadLine(f, line, 256);
while (line[0] != '$') {
nset = (struct shapeset *)
calloc(1,sizeof(struct shapeset));
nset->index = GetResidues(line, nset);
ReadLine(f, line, 256);
GetContact(line, nset);
if (!*set) *set = nset;
else {
struct shapeset *current = *set;
while (current->next) current = current->next;
current->next = nset;
}
i = ReadLine(f,line,256);
}
fclose(f);
}
void PrintCount( struct shapeset *shapes, FILE *output )
{
fprintf( output, "%d %d\n", shapes->index, shapes->seqcount );
if ( shapes->next ) PrintCount( shapes->next, output );
}
void PrintSeq( struct seqlist *seq, FILE *output )
{
fprintf( output, "%d ", seq->sequence );
if ( seq->next ) PrintSeq( seq->next, output );
}
void PrintKBMap( struct shapeset *shape, FILE *output )
{
if ( shape->slist ) {
fprintf( output, "%d ", shape->index );
PrintSeq( shape->slist, output );
fprintf( output, "\n" );
}
if ( shape->next ) PrintKBMap( shape->next, output );
}
void PrintKEMap( struct shapeset *shape, FILE *output )
{
fprintf( output, "%d %d\n", shape->index, shape->kmapseq );
if ( shape->next ) PrintKEMap( shape->next, output);
}
void MakeSeq( )
{
char inc = 1;
int i = 0;
while ( inc ) {
if ( Seq[i] ) {
Seq[i] = 0;
i++;
}
else {
Seq[i] = 1;
inc = 0;
}
}
}
void CleanSeq( )
{
int i;
for(i=0; i<SEQLENGTH; i++) Seq[i] = 0;
}
void CleanList( struct seqlist *sl )
{
if ( sl->next )
CleanList( sl->next );
free( sl );
}
void CleanShape( struct shapeset *shape)
{
shape->energy = 100;
if ( shape->slist ) CleanList( shape->slist );
shape->slist = NULL;
if ( shape->next ) CleanShape( shape->next );
}
/////////////////////////////////////////////////////////////////
// Symmetry detection functions
/////////////////////////////////////////////////////////////////
int IsSame(int X1s[], int Y1s[], int X2s[], int Y2s[])
{
int res = 1;
int i;
for (i=0; i<SEQLENGTH; i++) {
if (X1s[i] != X2s[i]) {res = 0; break;}
if (Y1s[i] != Y2s[i]) {res = 0; break;}
}
return res;
}
int IsXYSymmetry(int X1s[], int Y1s[], int X2s[], int Y2s[])
{
int res = 1;
int i;
for (i=0; i<SEQLENGTH; i++) {
if (X1s[i] != Y2s[i]) {res = 0; break;}
if (Y1s[i] != X2s[i]) {res = 0; break;}
}
return res;
}
int IsRXYSymmetry(int X1s[], int Y1s[], int X2s[], int Y2s[])
{
int res = 1;
int i;
for (i=0; i<SEQLENGTH; i++) {
if (X1s[i] != -Y2s[i]) {res = 0; break;}
if (Y1s[i] != -X2s[i]) {res = 0; break;}
}
return res;
}
int IsXSymmetry(int X1s[], int Y1s[], int X2s[], int Y2s[])
{
int res = 1;
int i;
for (i=0; i<SEQLENGTH; i++) {
if (X1s[i] != X2s[i]) {res = 0; break;}
if (Y1s[i] != -Y2s[i]) {res = 0; break;}
}
return res;
}
int IsYSymmetry(int X1s[], int Y1s[], int X2s[], int Y2s[])
{
int res = 1;
int i;
for (i=0; i<SEQLENGTH; i++) {
if (X1s[i] != -X2s[i]) {res = 0; break;}
if (Y1s[i] != Y2s[i]) {res = 0; break;}
}
return res;
}
int DNinty(int X1s[], int Y1s[], int X2s[], int Y2s[])
{
int res = 1;
int i;
for (i=0; i<SEQLENGTH; i++) {
if (X1s[i] != -Y2s[i]) {res = 0; break;}
if (Y1s[i] != X2s[i]) {res = 0; break;}
}
return res;
}
int DReverse(int X1s[], int Y1s[], int X2s[], int Y2s[])
{
int res = 1;
int i;
for (i=0; i<SEQLENGTH; i++) {
if (X1s[i] != -X2s[i]) {res = 0; break;}
if (Y1s[i] != -Y2s[i]) {res = 0; break;}
}
return res;
}
int DMNinty(int X1s[], int Y1s[], int X2s[], int Y2s[])
{
int res = 1;
int i;
for (i=0; i<SEQLENGTH; i++) {
if (X1s[i] != Y2s[i]) {res = 0; break;}
if (Y1s[i] != -X2s[i]) {res = 0; break;}
}
return res;
}
int IsRotation(int X1s[], int Y1s[], int X2s[], int Y2s[])
{
if (DNinty(X1s, Y1s, X2s, Y2s) ||
DReverse(X1s, Y1s, X2s, Y2s) ||
DMNinty(X1s, Y1s, X2s, Y2s)
) return 1;
return 0;
}
int IsSymmetry( struct shapeset *shape1, struct shapeset *shape2 )
{
int XReverse[SEQLENGTH];
int YReverse[SEQLENGTH];
int i;
for (i=0; i<SEQLENGTH; i++) {
XReverse[i] = shape2->Xs[SEQLENGTH-1-i];
YReverse[i] = shape2->Ys[SEQLENGTH-1-i];
}
if ( IsXYSymmetry(shape1->Xs, shape1->Ys, shape2->Xs, shape2->Ys) ||
IsSame(shape1->Xs, shape1->Ys, XReverse, YReverse) ||
IsXYSymmetry(shape1->Xs, shape1->Ys, XReverse, YReverse) ||
IsRXYSymmetry(shape1->Xs, shape1->Ys, XReverse, YReverse) ||
IsXSymmetry(shape1->Xs, shape1->Ys, XReverse, YReverse) ||
IsYSymmetry(shape1->Xs, shape1->Ys, XReverse, YReverse) ||
IsRotation(shape1->Xs, shape1->Ys, XReverse, YReverse)
) return 1;
return 0;
}
//////////////////////////////////////////////////////////////
// Compute NEC energy and mapping sequences to structure.
//////////////////////////////////////////////////////////////
/********************************************************
* Compute the NEC energy
********************************************************/
int ComputeNECEnergy( struct shapeset *shapes )
{
int E = 0;
struct contact *cs = shapes->clist;
if ( Seq[cs->i-1] && Seq[cs->j-1] )
E += HHENERGY;
else if ( Seq[cs->i-1] || Seq[cs->j-1] )
E += HPENERGY;
while ( cs->next ) {
cs = cs->next;
if ( Seq[cs->i-1] && Seq[cs->j-1] )
E += HHENERGY;
else if ( Seq[cs->i-1] || Seq[cs->j-1] )
E += HPENERGY;
}
return -E;
}
int FindMinValue( struct shapeset *shape )
{
int MinE, TmpE;
MinE = ComputeNECEnergy(shape);
shape->energy = MinE;
while ( shape->next ) {
shape = shape->next;
TmpE = ComputeNECEnergy(shape);
shape->energy = TmpE;
if (TmpE<MinE) MinE = TmpE;
}
return MinE;
}
void AddRecord(struct shapeset *shape, int sequence, int energy)
{
struct seqlist *sl;
struct seqlist *news =
(struct seqlist*)calloc(1,sizeof(struct seqlist));
news->sequence = sequence;
news->energy = energy;
if ( !shape->slist ) shape->slist = news;
else {
sl = shape->slist;
while ( sl->next ) sl = sl->next;
sl->next = news;
}
shape->seqcount ++;
}
void FindShape( struct shapeset *shape, int MinE, int Sequence )
{
struct shapeset *themini = NULL;
int i = 0;
if ( shape->energy == MinE )
themini = shape;
while ( shape->next ) {
shape = shape->next;
if ( shape->energy == MinE ) {
if ( !themini ) themini = shape;
else {
themini = NULL;
break;
}
}
}
if ( themini ) AddRecord( themini, Sequence, MinE );
}
void MapSequence( struct shapeset *shapes )
{
int seqval = 0;
int miniE;
CleanSeq( );
CleanShape( shapes );
miniE = FindMinValue( shapes );
FindShape( shapes, miniE, seqval);
while ( seqval<SEQMAX ) {
seqval++;
MakeSeq();
miniE = FindMinValue( shapes );
FindShape( shapes, miniE, seqval );
}
}
////////////////////////////////////////////////////////////
// Compute Kleinberg fitness value and brutal force mapping
////////////////////////////////////////////////////////////
int GetExposedSurface( int X, int Y )
{
if ((1==X || 4==X) &&
(1==Y || 4==Y) )
return 2;
if ( 1==X || 4==X ||
1==Y || 4==Y )
return 1;
return 0;
}
double CompKEnergy( struct shapeset *shapes )
{
int hydrop = 0;
int surp = 0;
int i;
struct contact * cl = shapes->clist;
for (i=0; i<SEQLENGTH; i++)
surp += Seq[i] * GetExposedSurface(shapes->Xs[i],
shapes->Ys[i] );
while (cl) {
hydrop += Seq[cl->i-1] * Seq[cl->j-1];
cl = cl->next;
}
return Alpha*hydrop + Belta*surp;
}
void FindMinSequence( int sequence, struct shapeset *shapes ) {
double energy;
energy = CompKEnergy( shapes );
if ( !shapes->slist ) {
shapes->slist =
( struct seqlist* )calloc( 1,sizeof( struct seqlist ) );
shapes->slist->sequence = sequence;
shapes->energy = energy;
} else if ( energy == shapes->energy ) {
struct seqlist *sl =
( struct seqlist* )calloc( 1,sizeof( struct seqlist ) );
sl->sequence = sequence;
sl->next = shapes->slist;
shapes->slist = sl;
} else if ( energy < shapes->energy ) {
CleanList( shapes->slist );
shapes->slist =
( struct seqlist* )calloc( 1,sizeof( struct seqlist ) );
shapes->slist->sequence = sequence;
shapes->energy = energy;
}
}
void KMinMap ( struct shapeset *shapes )
{
int seqval = 0;
struct shapeset *shape = shapes;
CleanSeq( );
CleanShape( shapes );
while( shape ) {
FindMinSequence ( seqval, shape );
shape = shape->next;
}
for ( seqval = 1; seqval<SEQMAX; seqval++ ) {
MakeSeq( );
shape = shapes;
while( shape ) {
FindMinSequence ( seqval, shape );
shape = shape->next;
}
}
}
//////////////////////////////////////////////////////////
//Kleinberg Algorithm (graph Mini-Cut) to find the
//sequence(s) for particular structure.
//////////////////////////////////////////////////////////
int MakeGraph( struct shapeset *shape,
graph &G,
node &source,
node &sink,
list<node> &snodelist,
list<node> &cnodelist,
list<edge> &edgelist,
edge_array<int> &weight) {
int i, len;
list<int> cap;
int X;
int Y;
source = G.new_node();
sink = G.new_node();
for (i=0; i<SEQLENGTH; i++) {
node n = G.new_node();
snodelist.append(n);
int sf = Belta*GetExposedSurface( shape->Xs[i],
shape->Ys[i] );
if (sf > 0 ) {
edge e = G.new_edge(n, sink);
edgelist.append(e);
cap.append(sf);
}
}
struct contact *c = shape->clist;
i = 0;
while ( c ) {i ++; c = c->next;}
c = shape->clist;
Y = abs( Alpha*i )+1;
X = abs( Alpha );
while ( c ) {
node n = G.new_node();
cnodelist.append(n);
edge e = G.new_edge(source, n);
edgelist.append(e);
cap.append(X);
edge e1 = G.new_edge(n,
snodelist.contents(snodelist.get_item(c->i-1)));
edge e2 = G.new_edge(n,
snodelist.contents(snodelist.get_item(c->j-1)));
edgelist.append(e1);
edgelist.append(e2);
cap.append(Y);
cap.append(Y);
c = c->next;
}
weight.init(G);
len = edgelist.length();
for (i=0; i<len; i++) {
weight[edgelist.contents(edgelist.get_item(i))]
= cap.contents(cap.get_item(i));
}
return Y;
}
void MarkGraph(graph G, node source,
edge_array<int> weight,
edge_array<int> res,
const list<node> &snodelist,
list<node> &partition,
int Max)
{
list<node> marked;
list<node> queue;
node current;
int i, len;
queue.append(source);
while (!queue.empty()) {
current = queue.pop();
marked.append(current);
list<edge> outedge = G.out_edges(current);
len = outedge.length();
for (i=0; i<len; i++) {
edge e = outedge.contents(outedge.get_item(i));
if ((weight[e] == Max)
|| (res[e] < weight[e] && res[e] > 0)) {
node n = G.target(e);
if (marked.search(n) == NULL) {
queue.append(n);
if (snodelist.search(n) != NULL)
partition.append(n);
}
}
}
}
}
int FindSeq(const list<node> &snodelist, const list<node> &partition )
{
int i;
int s=0;
for (i=0; i<SEQLENGTH; i++) {
node n = snodelist.contents(snodelist.get_item(i));
if (partition.search(n) != NULL) s += _Pow_int(2, i);
}
return s;
}
void KMinMapE( struct shapeset *shape )
{
int flow, max;
graph G;
node source;
node sink;
list<node> snodelist;
list<node> cnodelist;
list<edge> edgelist;
edge_array<int> weight;
edge_array<int> res;
list<node> marks;
while ( shape )
{
max = MakeGraph(shape, G, source, sink,
snodelist, cnodelist, edgelist, weight);
res.init(G);
flow = MAX_FLOW(G,source, sink, weight, res);
MarkGraph(G, source, weight, res, snodelist, marks, max);
shape -> kmapseq = FindSeq(snodelist, marks);
G.clear();
snodelist.clear();
cnodelist.clear();
edgelist.clear();
marks.clear();
shape = shape->next;
}
}
/////////////////////////////////////////////////////////////
// main
/////////////////////////////////////////////////////////////
int main(int argc, char **argv)
{
struct shapeset *shapes = NULL;
FILE *NECRes = fopen( "nec.txt", "w" );
FILE *KBRes = fopen( "kbr.txt", "w" );
FILE *KERes = fopen( "ker.txt", "w" );
ReadConformation( "shapes.txt", &shapes );
MapSequence( shapes );
PrintCount( shapes, NECRes );
KMinMap( shapes );
PrintKBMap( shapes, KBRes );
KMinMapE( shapes );
PrintKEMap( shapes, KERes );
return 0;
}
Jian Zhang
2000-06-18